

 For all latest articles, follow on Google News
For all latest articles, follow on Google NewsMolecular mechanism of metformin for type 2 diabetes
AUTHORS Dr. Rashidul Haque: Former Pro-Vice Chancellor, Varendra University, Rajshahi, Bangladesh; Former Professor, Emory University School of Medicine, Atlanta, USA; Former Associate Professor, Department of Zoology, University of Rajshahi, Bangladesh. Dr. Sultana Rajia: Assistant Professor, Center for Interdisciplinary Research (CIR), Varendra University, Rajshahi, Bangladesh.
Introduction
Metformin, a biguanide group of drugs, is currently the most prescribed drug for the treatment of type 2 diabetes mellitus in humans and has been used since the 1960s. It is also used to treat polycystic ovary syndrome (PCOS). A natural source of biguanide drug is a herbaceous plant called the French lilac, Galega officinalis. Gelegine, derived from that plant, is a natural substance that was used 100 years ago as an herbal medicine in medieval Europe but was found to be toxic to humans. Around the same time, two synthetic derivatives of gelegine, metformin and phenformin, were tested for three decades, while it was approved for human use in the late fifties, and it only received approval by the U.S. Food and Drug Administration for type 2 diabetes in 1994. Unlike most modern drugs, metformin, as it was a natural herbal product, was not designed to target any specific cell signaling pathway. The cellular response to this drug at the molecular level was also unknown. Even after 60 years of its clinical use, a complete picture of metformin’s pharmacological actions at the molecular level has not yet emerged. Metformin is generally well tolerated medicine. However, common adverse effects include diarrhea, nausea, abdominal pain, and lactic acidosis, a condition of an increased lactate production, that requires medical attention. Metformin is on the World Health Organization’s List of Essential Medicines (WHO, 2019). In this review, we have highlighted the unique properties of metformin in lowering glucose production in the body and explained how its benefits are likely to be caused by a variety of molecular mechanisms.
Metformin decreases intestinal glucose absorption, decreases hepatic glucose production, and enhances insulin sensitivity by increasing glucose utilization in the peripheral tissues, such as cardiac or skeletal muscle cells and adipose tissue. However, because of its positive charge and low lipophilicity, rapid passive diffusion of metformin through cell membranes is unlikely. Therefore, it requires an organic cation transporter (OCT) to enter and exit the cell. Seven such transporter proteins are involved in metformin trafficking in different cells of the body. For example, metformin enters intestinal enterocytes via OCT3 and ‘plasma membrane monamine transporter’ (PMAT) and exits via OCT1. However, metformin enters hepatocytes via OCT1 and OCT3 and exits via the ‘multidrug and toxin extrusion’ (MATE1) transporter, and it is released from the renal epithelial cells via OCT2 and MATE1/2. Also, metformin is not metabolized; it is cleared from the body by tubular secretion and excreted unchanged in the urine.
Metformin-mediated glucose-lowering effect begins in the intestine
Although the liver is recognized as a major site of metformin pharmacodynamics, several studies have demonstrated the role of the intestine in the blood glucose-lowering effect of metformin. Metformin does this by altering intestinal glucose absorption, with metformin increasing anaerobic glucose metabolism in enterocytes, resulting in reduced net glucose uptake and increased lactate delivery to the liver. Two transporter proteins are directly involved in the absorption of glucose in the intestine- glucose transporter 2 (GLUT2) and sodium-glucose linked transporter (SGLT)-1. GLUT2, a member of the glucose transporter family, is a bidirectional transporter. It helps in the uptake of glucose by the hepatocytes for the glycolysis and glycogenesis, and GLUT2 also regulates the release of glucose from the liver cells into the circulation during gluconeogenesis. GLUT2 is expressed in the renal tubular cells, liver cells, pancreatic beta cells and the small intestinal epithelial enterocytes. Glucose is absorbed from the intestinal lumen into the enterocytes through the cotransporter SGLT-1 protein or/and GLUT2 (controversial) located in the apical membrane of the enterocytes, whereas GLUT2 is always considered to provide basolateral exit. Mutations in GLUT2 or SGLT-1 genes have been shown to impair glucose absorption, indicating the importance of these two proteins in glucose absorption. Furthermore, the same response has been observed in the early response to orally administered metformin, i.e. metformin favors glucose-absorption and subsequent lowering of blood glucose levels. All GLUT family proteins are encoded by the solute carrier SLC2 gene family located in the human DNA.
Metformin may also impact on glucose-lowering effects in the intestine by increasing glucagon-like peptide-1 (GLP-1) secretion. Additionally, a fascinating gut-mediated mechanism for metformin action was found in rats, where a pathway links duodenal metformin exposure to suppression of hepatic glucose production, via the nucleus tractus solitarius and vagal efferents, through the AMP-activated protein kinase (AMPK), a serine/threonine-specific protein kinase, and GLP-1 receptor activation- a gut–brain–liver crosstalk.
At the molecular level, how metformin works in different cell types is still not very clear to the scientists. However, metformin is known to inhibit the mitochondrial respiratory chain (complex I), activates AMPK, inhibits glucagon-induced cyclic adenosine monophosphate (cAMP) stimulation, inhibits mitochondrial glycerophosphate dehydrogenase (GPD2), a key component of the glycerophosphate shuttle between the cytoplasm and the mitochondria, that results in decreased hepatic gluconeogenesis. Metformin also affects the gut microbiota.
What is gluconeogenesis?
Gluconeogenesis is a metabolic pathway by which glucose is produced from certain non-carbohydrate carbon substrates (such as glucogenic amino acids, lactic acid/lactate, pyruvate and glycerol) and this ubiquitous process is present in all living organisms, including bacteria, and other microorganisms. Gluconeogenesis is an energy intensive process, which consumes six ATP equivalents per molecule of glucose synthesized. Therefore, hepatocytes need to balance the demand for ATP with supply, with the latter primarily provided by mitochondria. In vertebrates, gluconeogenesis is mostly confined to the liver and occurs to a lesser extent in the renal cortex, muscle, intestine and in astrocytes of the brain. The liver uses lactate, glycerol and amino acids (especially alanine) as gluconeogenic precursors. On the other hand, kidneys use lactate, glutamine and glycerol. Gluconeogenesis is one of two primary processes – the other is the breakdown of glycogen (glycogenolysis) – that humans and many other animals use to maintain blood sugar levels, avoiding low levels (hypoglycemia).
Lactate is one of the many gluconeogenic sources available in liver cells. Lactate is converted to pyruvate by the enzyme lactate dehydrogenase (LDH). Glucose-6-phosphatase (G6Pase), fructose-1,6-bisphosphatase (FBPase) and phosphoenolpyruvate carboxykinase (PEPCK) are very important in gluconeogenesis proceseys, and also key targets of metformin. Various protein factors and hormones regulate the process of gluconeogenesis. Insulin and glucagon have the opposite effects: whereas, glucagon stimulates the process of gluconeogenesis in fasting condition, insulin prevents the excess blood glucose. Glucagon and insulin mainly act through some important transcription factors. The activation of glucagon hormone during fasting states stimulates the transcription factors CREB 1 (cAMP response element-binding protein), CBP (CREB-binding protein), CRTC2 (CREB-regulated transcription co-activator 2), FoxO1 (forkhead transcription factor) and FoxO6. These factors are involved in the transcription of G6Pase, FBPase and PEPCK genes, the most important genes regulating the process of gluconeogenesis. On the other hand, the activation of insulin in the presence of excess blood glucose inhibits FoxO1 and FoxO6 by phosphorylating them by protein kinase B (Akt). However, insulin resistance results in the failure of insulin to block those transcription factors, resulting in the continued gluconeogenesis despite high blood glucose levels. In this condition, metformin is able to inhibit the CREB-CBP-CRTC2 complex and FoxO6. For this reason, gluconeogenesis is an important target of type 2 diabetes therapy.
The site of cellular glucose production in the body is the liver, where four glucose-related metabolic processes, namely gluconeogenesis, glycogenolysis (conversion of glycogen to glucose), glycogen synthesis (conversion of glucose to glycogen) and glycolysis (conversion of glucose to pyruvate) ultimately determine the net glucose production in a cell. After overnight fasting, these four metabolic processes account for approximately 85 to 90 percent of cellular glucose production, with gluconeogenesis itself contributing approximately 50%. Therefore, gluconeogenesis has become a major target of metformin in glycemic control as a type 2 diabetes therapy in the body.
Mitochondrion and gluconeogenesis are the main targets of metformin in the liver
Metformin lowers blood glucose levels by reducing glucose production in the liver and by increasing glucose utilization in the muscles (especially the skeletal muscle). The most intensively studied mitochondrial action of metformin is the inhibition of Complex I of the mitochondrial respiratory chain, which suppresses ATP production. Therefore, the reduced proton gradient and the increased AMP/ATP and ADP/ATP ratios in the cytoplasm and the increased lactate/pyruvate ratio stimulate AMPK. Besides ATP production, other consequences of the respiratory chain inhibition such as changes in the nicotinamide adenine dinucleotide ratio (NAD+/NADH, also known as pseudohypoxia), may also contribute to the effects of metformin on gluconeogenesis. Whether higher (mmol/l) or lower (µmol/l) extracellular concentration of metformin does this suppression is still a matter of debate. Among the potential effectors of metformin in liver cells is the master regulator of glucose production, AMPK, which consists of α, β, and γ subunits. It has been established that AMPK is an important bioenergetic sensor and the cellular regulator of glucose, lipid and protein metabolism. Therefore, research focusing on AMPK has also been abundant. In 2021-22 itself, about five hundred research papers related to glucose and AMPK were published (https://www.ncbi.nlm.nih.gov/).
As mentioned, Metformin’s main target in the liver is to inhibit gluconeogenesis and this inhibitory effect of metformin on glucose production requires activation of the cellular energy sensor AMPK (Figure 1). The activation of AMPK is mediated by the phosphorylation of metformin-stimulated liver kinase B1 (LKB1), an upstream kinase of AMPK. Therefore, LKB-1 knockout mice abrogated the anti-hyperglycemic effect of metformin. Several studies have shown that the inhibitory effect on hepatic glucose production by metformin may be mediated by both AMPK-dependent and AMPK-independent mechanisms. As shown, a single mutation in the AMPK-mediated phosphorylation site of acetyl-CoA carboxylase (ACC1/2) strongly blocks the metformin-stimulated insulin sensitivity and glucose tolerance in diabetic rats, indicating the importance of AMPK activation in metformin activity.
Metformin exerts its effects in the liver by two mechanisms. First, metformin increases the amount and activity of adenosine monophosphate (AMP) within the cell (cytoplasm). Also, by inhibiting the activity of GPD2, metformin decreases NADH oxidation, decreases proton (H+) pumping or proton movement across the inter-mitochondrial membrane, inhibits overall ATP synthesis, and thus reduces the rate of oxygen consumption. Therefore, the reduced proton gradient and the increased AMP/ATP and ADP/ATP ratios in the cytoplasm, reduced NAD+/NADH ratio and the increased lactate/pyruvate ratio eventually activate AMPK. In addition, the increased AMP in the cell binds to a subunit of AMPK making it more sensitive for phosphorylation by LKB1. As shown, the activated AMPK increases hepatic insulin sensitivity and switches hepatocytes from anabolic pathways, such as gluconeogenesis, fatty acid and protein synthesis, to catabolic pathways, such as glycolysis and fatty acid oxidation, which consume less energy and restore energy balance. Consequently, the stimulated AMPK inhibits transcription factors including hepatocyte nuclear factor 4 (HNF4) and CRTC2 and the genes involved in gluconeogenesis, such as PEPCK, G6Pase and FBPase, and decreased glucose production as well as lipogenesis-like processes through the phosphorylation of two isoforms of acetyl-CoA carboxylase (ACC 1/2) at equivalent serine residues. This results in the decreased diacylglycerol content in the liver and improved insulin sensitivity.
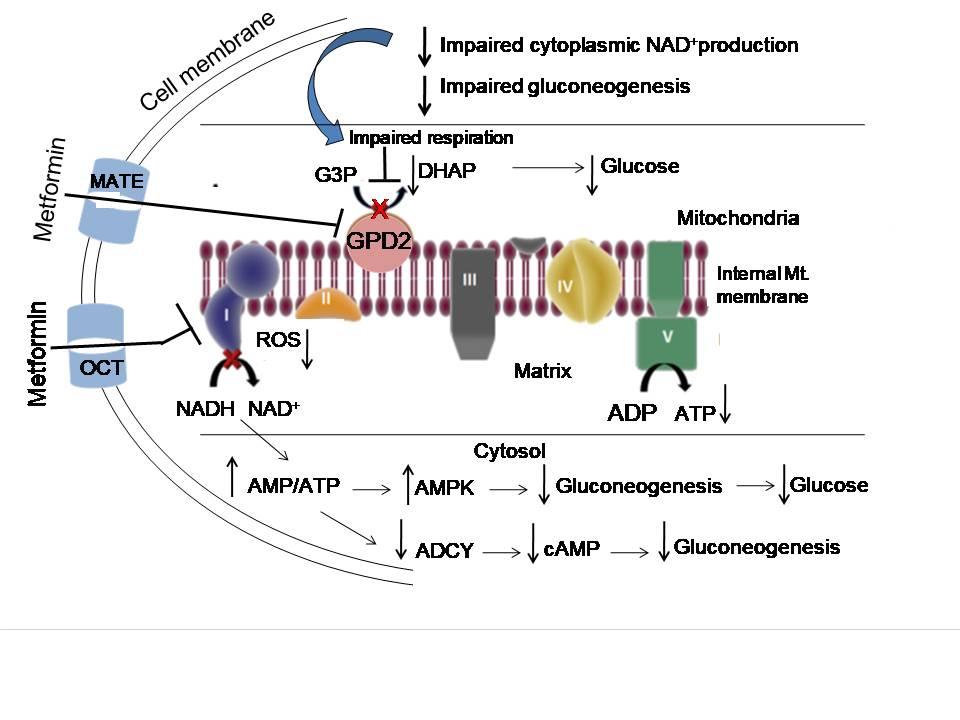
Alternatively, metformin is able to activate AMPK without inhibiting the complex-I of the mitochondrial respiratory chain and without altering the AMP/ATP ratio in the cytoplasm. Those alternative mechanisms include the decreased production of cAMP by inhibiting the enzyme adenylate cyclase, the increased secretion of GLP-1 in the enterocytes, and the decreased activity of the dipeptidyl peptidase-4 (DPP-4), an enzyme that inactivates GLP-1. GLP-1 peptide, a member of a family of hormones called incretins, plays a unique role in regulating the glucose levels in type 2 diabetes, increasing insulin secretion from pancreatic beta-cells and inhibiting glucagon secretion. Cells lining the small intestine (called enteroendocrine ‘L-cells’) are the main source of GLP-1, although it is secreted in smaller amounts from the pancreas and the central nervous system. In addition, the increased AMP level decreases the availability of cAMP in cells by inhibiting the adenylate cyclase activity, thus inhibiting the cAMP-PKA pathway, which eventually inhibits the PKA-mediated CRTC2:CREB activation. As previously mentioned, the CRTC2:CREB complex (transcription factor complex) is directly involved in the expression of three gluconeogenesis-activating genes, PEPCK, G6Pase and FBPase. The AMPK-mediated phosphorylation of the cAMP specific 3′,5′-cyclic phosphodiesterase 4B (PDE4B) is another way to trigger cAMP breakdown.
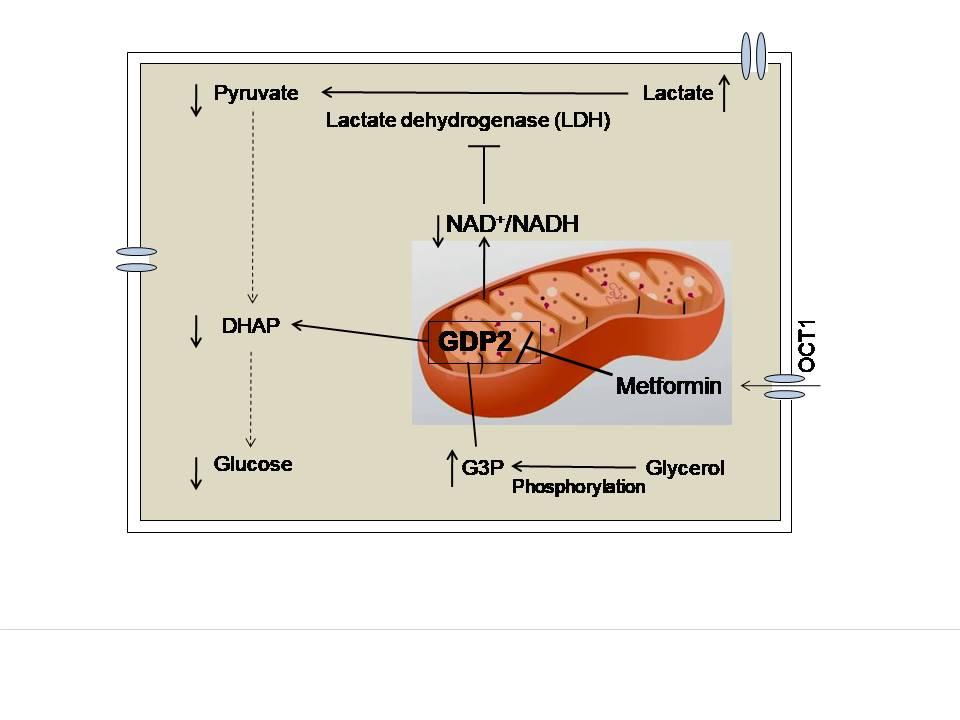
An important different target of metformin’s therapeutic effects is the mitochondrial GPD2 enzyme. In the process of lipolysis, white adipose tissue produces glycerol and nonesterified fatty acids (NEFA), both of which stimulate gluconeogenesis in the liver. NEFA enters mitochondria to form acetyl-CoA. It can directly participate in gluconeogenesis by converting pyruvate to oxaloacetate by pyruvate carboxylase. On the other hand, glycerol itself is a gluconeogenic substrate that is converted to glycerol-3-phosphate and is further converted by GPD2 to dihydroxyacetone phosphate (DHAP). Metformin decreases the cellular NAD+/NADH ratio by inhibiting this catalytic reaction by GPD2, thereby inhibiting gluconeogenesis specifically from glycerol and lactate (Figure 2). It should be noted here that there is still a disagreement among scientists as to whether the metformin-mediated inhibition of GPD2 enzyme decreases or increases the amount of NAD+ in the cells, or does not affect the NAD+/NADH ratio at all.
Yet, another pathway of the metformin-mediated AMPK activation other than the mitochondria has been identified, which is the lysosomal pathway. Metformin does this by binding to the PEN2 (presenilin enhancer 2) protein, a subunit of the gamma-secretase (γ-secretase) enzyme. Metformin-PEN2 binds together with ATP6AP1 to inhibit the lysosomal proton pump v-ATPase and activate AMPK. However, the metformin-mediated AMPK activation through the lysosomal pathway has not been described here in detail.
Metformin and insulin sensitivity
Besides suppressing gluconeogenesis or glucose production in the liver, metformin increases insulin sensitivity, especially in the skeletal muscle and adipocytes, and increases fatty acid oxidation. Our knowledge of the insulin-stimulated glucose transport has advanced with the understanding of insulin-regulatable glucose transporter protein or glucose transporter 4 (GLUT4) at the molecular level and also its trafficking mechanism from the cytosol to the cell membrane. Muscle cells take up about 80% of the total glucose present in the body. However, decreased GLUT4 protein expression or defects in the translocation of GLUT4 from the cytosol to the membrane have been identified as a major cause of insulin resistance. In human cells, GLUT4 encoded by the SLC2A4 gene is predominantly expressed in skeletal muscle and adipose tissue, but small amounts of GLUT4 are also found in the brain, kidney, intestine and liver.
GLUT4-mediated transmembrane glucose transport is the rate-limiting step in peripheral glucose utilization. A 70% decrease in GLUT4 protein expression and a 72% reduction in insulin-stimulated glucose transport resulted from the adipose-specific genetic knockout of GLUT4. In addition to genetic defects, excessive fat- and sugar-rich diet, obesity, vitamin D deficiency, high blood pressure, excess cholesterol, and certain medications (such as corticosteroids) can also cause insulin resistance. Metformin is a widely prescribed insulin-sensitizing agent in the current clinical use. The experimental studies have shown that the metformin-mediated improvement in insulin sensitivity is associated with several mechanisms, including the increased insulin receptor (Ins-R) activity, increased glycogen synthesis, and enhanced GLUT4 expression and activity. In human adipocytes, 24h incubation with metformin significantly increased glucose uptake, GLUT4 mRNA expression, and GLUT4 content in the adipocyte plasma membrane. In addition, the metformin-mediated AMPK activation increases GLP-1 secretion from the pancreas, increases GLUT4 translocation (from the cytosol to the cell membrane), and lowers plasma glucose levels. Also, the metformin-induced AMPK activation decreased the transcriptional repressor histone deacetylase 5 (HDAC5), with a known association with the GLUT4 gene, resulting in increased GLUT4 expression in human myotubes. Additionally, a number of microRNAs have been reported to regulate insulin sensitivity and GLUT4 expression, and their levels were shown to be altered in insulin resistance (IR) conditions. Moreover, changes in gut microbiota density may also be an additional target for metformin.
Metformin maintains the insulin signaling pathway by increasing insulin secretion and the activation of Ins-R, a transmembrane receptor that belongs to the large class of the receptor tyrosine kinase, allowing glucose to enter cells more easily. Insulin facilitates the transport of glucose from blood into cells, thereby reducing blood glucose (blood sugar). Insulin does this by activating its own receptor (Ins-R) and certain activators in the cell, and it also has to follow a specific signaling pathway (Figure 3). Both insulin and insulin growth factor, IGF-1, follow the same signaling pathway. Binding insulin to Ins-R triggers autophosphorylation of the tyrosine residues, with each subunit phosphorylating its partner. As a result, the insulin receptor regains its activation and immediately activates ‘insulin receptor substrates (IRS-1, IRS-2)’ through phosphorylation. The activated IRS-1 starts signal transduction pathway and binds to phosphoinositide 3-kinase (PI3K) and gets phosphorylated. PI3K catalyzes conversion of PIP2 (phosphatidylinositol 4,5-bisphosphate) molecules to PIP3 (phosphatidylinositol 3,4,5-trisphosphate). PIP3 is an important signaling molecule, which in turn activates several other kinases, particularly PDPK (phosphoinositide-dependent kinase)-1 and protein kinase B (PKB, also known as Akt). PDPK-1 is a master kinase which is crucial for the activation of PKB. To facilitate the influx of glucose into the cell, PKB, with the help of vesicles and SNARE proteins, translocates the glucose transporter (GLUT4) from the cytoplasm to the cell membrane and establishes its specific location on the membrane.
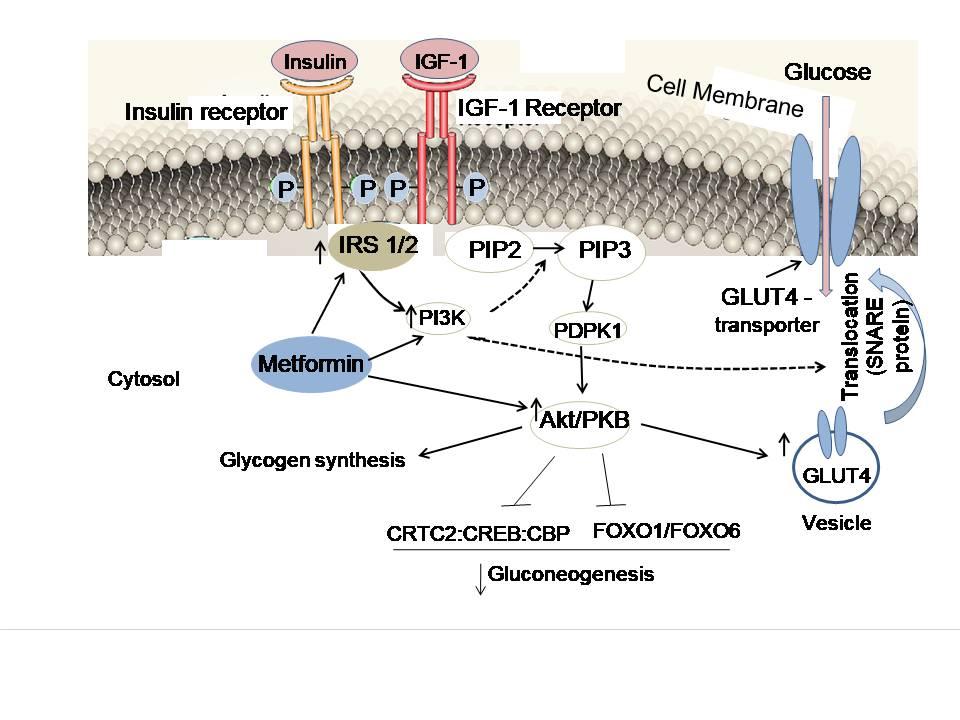
As mentioned earlier, if the secretion of insulin decreases or the efficiency of the insulin receptor declines, glucose does not reach its destination. As a result, blood glucose levels rise, resulting in diabetes or hyperglycemia, along with high blood pressure. The IRS-PI3K-PKB signaling has been described as a crucial pathway for the development of IR. It has been observed that the expression of PI3K, PKB and GLUT4 is decreased in various tissues of diabetic patients, but they returned to their normal expression level with metformin administration. In human hepatocytes and Huh7 cell cultures, metformin (1μg/ml) increased Ins-R tyrosine phosphorylation by 78% within 30 min, along with the increased stimulation of IRS-2. In addition, as reported in the insulin-stimulated granulosa-luteal cells, metformin increases both the mRNA and protein synthesis levels of IRS-1, activates the Akt enzyme through PI3K, and transports GLUT-4 vesicles from the cytosol to the cell membrane. Inositol-5-phosphatase 2 with Src homologous domain 2 (SHIP2) is overexpressed in IR tissues. However, metformin by inhibiting the activity of SHIP2 increases cellular glucose uptake (Polianskyte-Prause et al. 2019). SHIP2 normally represses PI3K-mediated insulin signaling. Also, the metformin-stimulated AMPK activation and increased activity of GLUT4 transporter protein increases cellular glucose uptake.
Conclusion
Metformin is a miracle drug with multiple sites of action and multiple molecular mechanisms to control diabetes, while increasing the insulin sensitivity in peripheral tissues. Central to these mechanisms is the activation of the AMPK by inhibiting the mitochondrial respiratory chain complex I in the liver, increasing AMP/ATP ratio, lowering cAMP level in the cytoplasm, thus reducing the glucose production by inhibiting gluconeogenesis. Additionally, metformin acts on the gut to increase glucose utilization, enhances GLP-1 secretion and alters the gut microbiome. Also, the metformin-mediated induction of GLUT4 expression and its enhanced translocation to the plasma membrane improves insulin sensitivity. Despite decades of research, the curiosity about the metformin’s mechanism of action at the molecular level has not ended. In addition to improving the glucose homeostasis, metformin may also reduce the risk of developing cancers. Studies have shown that metformin can reduce the risk of developing pancreatic cancer and tumors by up to 60 percent. Besides, the use of metformin has also increased significantly in the treatment of neurodegenerative diseases, obesity, polycystic ovary syndrome and others. Lowering blood levels of low-density lipoprotein (LDL) and triglycerides, metformin’s effect in the prevention of heart disease to a certain degree has also been reported. Yet, more research is needed to understand the pleiotropic effects of metformin at the cellular and molecular mechanisms.

{kind=link}